Got junk sequences in your sequencing FASTQs?
Many experiments are riddled with repetitive sequences which are of no
value to the scientific question you are asking. Some examples include
ribosomal RNA and hemoglobin. DASH (1) is a CRISPR-Cas9
next-generation sequencing (NGS) technology which depletes abundant
sequences, increasing coverage of your sequences of interest. ![]()
In the lab, you take final NGS libraries ready for sequencing, and combine them with pre-programmed Cas9 guide RNAs (gRNAs) which cut up your repetitive sequences into small fragments. You then size select and amplify your remaining DNA, which should contain most or only your sequences of interest.
DASHit (2) is the software that designs Cas9-gRNAs to target your particular experiment.
For more details on the wet lab side, check out our actual protocol.(3)
- Gu, W. et al. Depletion of Abundant Sequences by Hybridization (DASH): using Cas9 to remove unwanted high-abundance species in sequencing libraries and molecular counting applications. Genome Biology 17, 41 (2016).
-
Dynerman, D and Lyden, A. et al.
Preprint soon! - Lyden, A. et al. DASH Protocol V.4.
Installing
Please visit the GitHub repository for instructions on how to install DASHit.
The Idea
In order for DASH to be maximally effective, we want to pick the fewest guides which hit the largest possible number of sequences that you want depleted. The best way to find the over-abundant sequences in your data is to look at it!
DASHit uses reads from a preliminary, low-depth sequencing of your samples to identify abundant CRISPR-Cas9 cut sites. It has a filtering function for the guides it identifies, which allows you to specify on and off target sequences, and filter based on GC content and secondary structure. It then computes an optimized guide list by producing a set number of guides which hit the largest number of sequences in the preliminary data.
Next, we’ll describe the DASHit pipeline, or, you can skip ahead to a real example.
For even more detail, please see the DASHit paper (2).
DASHit Pipeline
DASHit is a collection of several tools you can use to design gRNAs that target the sequences you want.
INPUT: A FASTQ file input.fastq containing the abundant sequences you want to target for depletion.
NOTE: We usually subsample
input.fastqdown to several hundred thousand reads withseqtkbefore proceeding.
- Use seqtk (or another tool) to convert the input to FASTA format
seqtk seq -A input.fastq > input.fasta - Trim adaptor sequences using cutadapt (or another tool). This will avoid designing a guide which cuts your adaptor sequence, rendering your library unsequencable.
cutadapt -a AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC -o cut-input.fasta input.fasta The above is an example command for trimming adapters from NEBNext paired-end libraries. You will need to change the adapter sequence if you used a different library prep method.
The above is an example command for trimming adapters from NEBNext paired-end libraries. You will need to change the adapter sequence if you used a different library prep method. - Run
crispr_sites -rto find candidate Cas9-gRNAs in your input.cat cut-input.fasta | crispr_sites -r > input_sites_to_reads.txt Each line of
Each line of input_sites_to_reads.txtcontains a 20-mer Cas9 guide RNA found ininput.fasta, together with a list of reads that 20-mer appeared in -
(Optional, but you’ll probably want this) You may want to restrict your gRNAs to a particular ontarget region, and also disqualify guides that hit some offtarget region.
- Obtain FASTA files of your ontarget and offtarget regions
- Run
crispr_sitesto find ontarget and offtarget gRNAscat ontarget.fasta | crispr_sites > ontarget_sites.txt cat offtarget.fasta | crispr_sites > offtarget_sites.txt For the ontarget and offtarget files, run crispr_siteswithout the-rflag.
- Filter your candidate guide RNAs, removing those with low-quality structure (GC content, homopolymer, dinucleotide repeats, and hairpins). Optionally remove all guides RNAs present in
offtarget_sites.txt, and those not present inontarget_sites.txtdashit_filter --gc_freq_min 5 --gc_freq_max 15 --ontarget ontarget.txt --offtarget offtarget.txt input_sites_to_reads.txt > input_sites_to_reads_filtered.txt Run dashit_filter --helpfor an explanation of the quality filtering and to learn how to change quality thresholds. ontarget and offtarget filtering require port 8080 to be available on your computer. - Find 300 guides that hit the largest number of reads
optimize_guides input_sites_to_reads_filtered.txt 300 1 > guides.csv You should experiment with different numbers of guides the number of times to cover each readoption, here set to 1, is how many guides need to hit a read ininput.fastabefore that read is considered covered. In principle you could use this for additional redundancy, but in practice we’ve never needed anything other than 1 - Examine
guides.csvto see how we did. Open this file in Excel and plot the last column. This plot will show the cumulative number of reads hit ininput.fastathe designed guides hit. You can use this plot to pick the number of guides to use: look for the “elbow” in the plot, and notice where diminishing returns on the number of guides kicks in.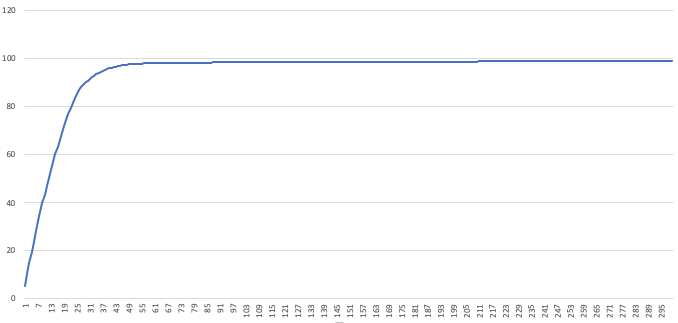 We see around 50 guides are already covering almost 100% of our
We see around 50 guides are already covering almost 100% of our input.fasta. Ordering 300 guides would be overkill, in this case - Score how well your guides do against
input.fasta- this should match the plot fromguides.csv.score_guides guides.csv input.fasta score_guidesis mainly useful to estimate how an existing guide set would perform against a new set of samples. You might not have to design new guides!
OUTPUT: guides.csv, your DASH guides!
![]() The contrib directory contains many useful scripts for automating parts of this workflow. For instance, these scripts will convert your
The contrib directory contains many useful scripts for automating parts of this workflow. For instance, these scripts will convert your guides.csv into a spreadsheet ready to place an order.
Example
For an example on real data, see here.